En los últimos días, la comunidad científica internacional ha alcanzado un logro crucial para la seguridad sanitaria global: la obtención de la secuencia genómica completa del letal Orthohantavirus andesense (conocido como virus Andes o ANDV). Este monumental esfuerzo de biología molecular acaba de ocurrir en respuesta a un aterrador brote mortal a bordo de un crucero transatlántico. Al descifrar la totalidad del código genético de este patógeno —el único hantavirus con capacidad demostrada de transmisión de persona a persona—, los investigadores han podido rastrear su origen exacto y comprender cómo se ha estado propagando silenciosamente entre los pasajeros.
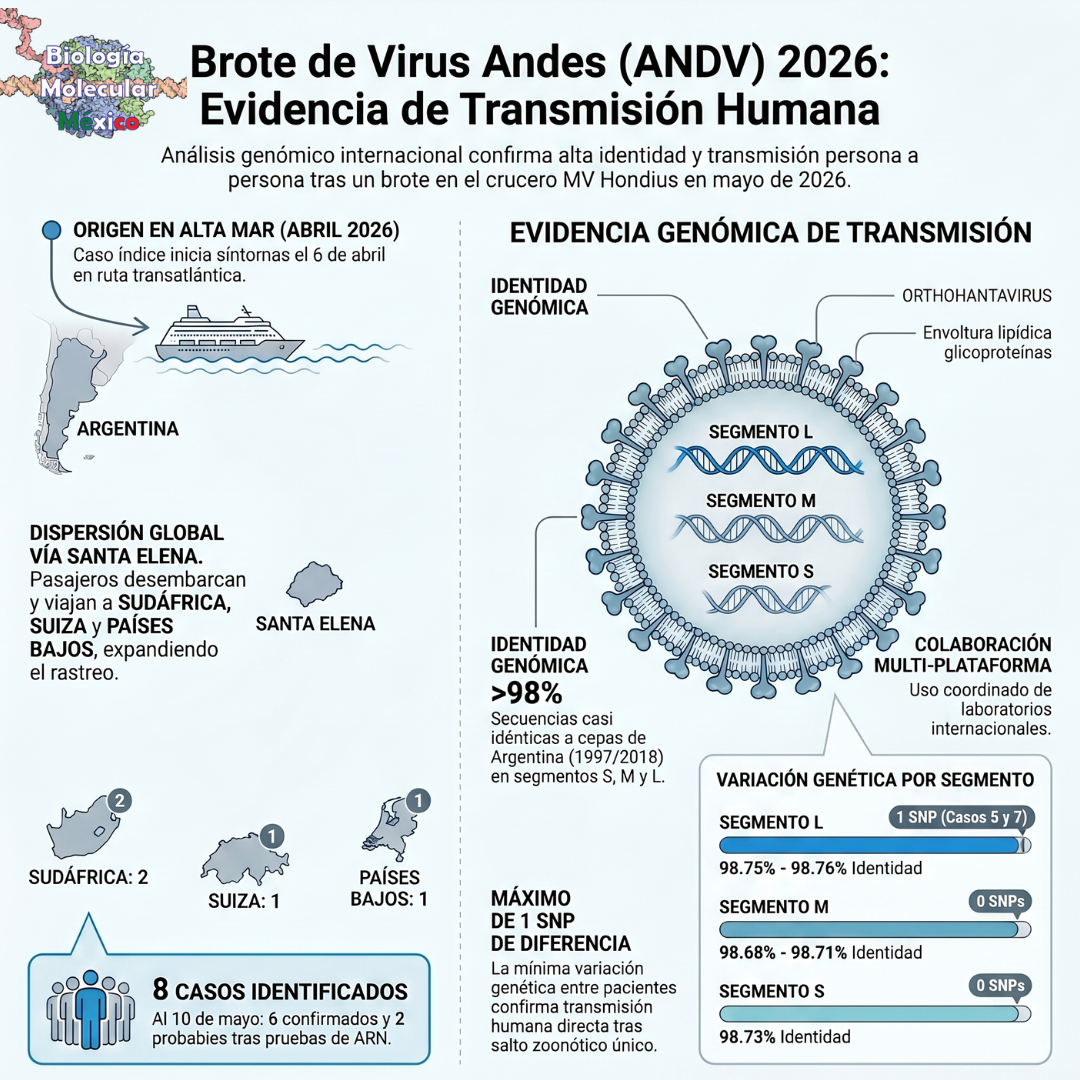
La cacería del genoma completo y el caso suizo La urgencia por secuenciar el virus ha comenzado luego de que el crucero MV Hondius, que zarpó desde Ushuaia (Argentina) a principios del mes pasado, reportara la trágica muerte de su caso índice y, posteriormente, de su esposa y otros pasajeros que desembarcaron en distintos continentes. Al dispersarse los infectados por el mundo, la respuesta ha exigido una colaboración inmediata en estos últimos días entre laboratorios de Sudáfrica, Senegal, Suiza y los Países Bajos.

El hito que acaba de marcar un antes y un después en esta investigación ha ocurrido hace apenas tres días, el 8 de mayo. Ese día, un equipo de los Hospitales Universitarios de Ginebra y la Universidad de Zúrich ha logrado publicar con éxito la primera secuencia genómica completa del virus, obtenida a partir del “paciente 7”, un residente suizo que acababa de desembarcar del barco en la isla de Santa Elena antes de volar de regreso a su país. Este mapa genético completo está sirviendo ahora mismo como la piedra roseta para el resto del consorcio internacional.
¡GRACIAS POR LEER NUESTRAS NOTICIAS! ¿Nos invitas un cáfe? ☕


El arsenal tecnológico detrás de la secuenciación Para obtener el genoma completo del resto de los pacientes y confirmar la estructura del virus en tiempo récord, los científicos han desplegado esta semana un impresionante arsenal de biología molecular. Debido a que las muestras varían en calidad (algunas con cargas virales extremadamente bajas), los institutos han combinado múltiples estrategias de preparación, como la metagenómica no sesgada, la amplificación SISPA y enfoques de enriquecimiento basados en captura TWIST.
El código genético ha sido leído utilizando las plataformas de secuenciación de nueva generación más avanzadas del mercado, incluyendo tecnologías de Illumina, Element BioSciences y la secuenciación de lectura larga de Oxford Nanopore.
Ciencia abierta: El escrutinio de los datos “crudos” Tener el genoma completo armado es solo una parte del reto. Al usar diferentes máquinas y algoritmos, existe el riesgo de interpretar errores de secuenciación como mutaciones reales. Para garantizar la precisión absoluta de la secuencia completa de manera inmediata, la comunidad científica ha abogado por la transparencia total.
A través de foros especializados, investigadores independientes acaban de solicitar el acceso a los datos bioinformáticos originales y “crudos” (como los archivos deshumanizados .fastq.gz o los archivos de alineación .bam). En un esfuerzo sin precedentes, el consorcio ha estado compartiendo estas lecturas virales en tiempo real. Esto ha permitido a la comunidad global calibrar de inmediato sus flujos de trabajo (workflows), estandarizar los parámetros de referencia y filtrar los artefactos técnicos de las mutaciones genuinas.
Los secretos que acaba de revelar la secuenciación completa Tener acceso a la secuencia completa del Hantavirus Andes ha permitido a los biólogos moleculares analizar a detalle los tres segmentos de ARN que componen su genoma: S (Pequeño), M (Mediano) y L (Grande). Los hallazgos que se acaban de publicar son concluyentes y escalofriantes.
Al comparar los genomas completos de los diferentes pasajeros infectados, los científicos han descubierto que los segmentos S y M son absolutamente idénticos en todos los casos. El segmento L apenas ha mostrado dos minúsculos cambios de una sola letra (SNPs): una mutación en la posición C2139T en el paciente 5, y otra en la posición G576A en el paciente 7. Crucialmente, ambas han demostrado ser mutaciones “sinónimas”, lo que significa que no alteran en absoluto a las proteínas virales.
El genoma completo ha revelado una similitud superior al 98.6% con cepas del virus Andes que circularon en Argentina en 1997 y 2018. Epidemiológicamente, esta apabullante falta de diversidad genética a lo largo de todo el código viral acaba de confirmar la peor sospecha: el brote se originó a partir de una única exposición a un roedor sudamericano (un evento de derrame zoonótico), seguido por un contagio directo y letal de humano a humano dentro del confinamiento del barco.
Gracias a la rápida secuenciación genómica completa que se ha compartido esta misma semana en plataformas como Pathoplexus, hoy las autoridades sanitarias globales tienen en sus manos las herramientas exactas para afinar sus pruebas de diagnóstico y contener a uno de los virus más amenazantes de la naturaleza.