📝 RESUMEN
Un estudio bioinformático histórico liderado por el Chan Zuckerberg Biohub y EvolutionaryScale ha presentado el ESM Atlas, el mapa de biodiversidad estructural más grande del mundo, impulsado por el revolucionario modelo de lenguaje ESMC (ESM Cambrian) y el predictor ESMFold2. A diferencia de sistemas como AlphaFold, que dependen de Alineamientos de Secuencias Múltiples (MSA) lentos y costosos en cómputo, este nuevo paradigma demuestra que el aprendizaje no supervisado a gran escala permite a la IA internalizar un “modelo de mundo” físico y evolutivo directamente de las secuencias de aminoácidos. Con un atlas que abarca 6,800 millones de proteínas metagenómicas y 1,100 millones de estructuras 3D predichas, ESMFold2 ha demostrado en pruebas de laboratorio una precisión superior en el acoplamiento de anticuerpos y una tasa de éxito de hasta el 88% en el diseño de novo de fármacos inmunológicos, todo liberado bajo código abierto y sin restricciones comerciales.
1. Del Texto a la Biología: El Concepto de “Modelo de Mundo” 🌍📖
¡GRACIAS POR LEER NUESTRAS NOTICIAS! ¿Nos invitas un cáfe? ☕

La gran tesis del artículo es que las proteínas pueden leerse exactamente como un idioma. Las letras son los aminoácidos y las palabras son los pliegues funcionales.
- El Aprendizaje No Supervisado: Entrenando al modelo gigante ESMC con miles de millones de secuencias del árbol de la vida (incluyendo metagenómica profunda de océanos y suelos), la IA aprendió a predecir qué aminoácido seguía en una secuencia oculta.

- La Emergencia de la Física: Al dominar la “gramática” evolutiva, el modelo desarrolló una capacidad interna sorprendente: entender la termodinámica, los ángulos de torsión atómica y las fuerzas electrostáticas de las proteínas sin haber sido entrenado explícitamente para ello. Es un simulador digital de la bioquímica molecular.
2. La Arquitectura de ESMFold2: Velocidad Extrema y Módulos de Difusión ⚙️⚡
El paper desglosa un cambio radical en el algoritmo que deja atrás el procesamiento lento de los co-evolutivos tradicionales:
- Integración de Representaciones de ESMC: El modelo toma los mapas neuronales del modelo de lenguaje y los inyecta directamente en capas de plegamiento por parejas (PairFoldingLayer).

- Módulo de Difusión Truncada (Truncated Diffusion Sampling): Para generar las coordenadas tridimensionales de los átomos con precisión milimétrica, ESMFold2 utiliza un algoritmo de difusión matemática que refina la posición del esqueleto de la proteína paso a paso, estabilizando los enlaces químicos de forma óptima. Esto le permite predecir estructuras complejas en una fracción de segundo, permitiendo el procesamiento masivo de miles de millones de muestras.
3. El Triunfo Inmunológico: Superando a AlphaFold en Anticuerpos 🥊🛡️
Uno de los puntos más fuertes del artículo científico es la evaluación cara a cara con los modelos de Google DeepMind en el campo de las interacciones moleculares complejas.
- El Desafío de los Multímeros: Predecir cómo interactúan dos proteínas distintas (como un virus y un receptor celular) es sumamente difícil debido a la flexibilidad de las cadenas proteicas.
- Especificidad Anticuerpo-Antígeno: ESMFold2 demostró una precisión sin precedentes en la predicción de la zona de contacto (epitopo y paratopo) de los anticuerpos. Superó las métricas de AlphaFold 3 en benchmarks inmunológicos clave, convirtiéndose en la herramienta definitiva para la ingeniería de sueros y terapias biológicas dirigidas.
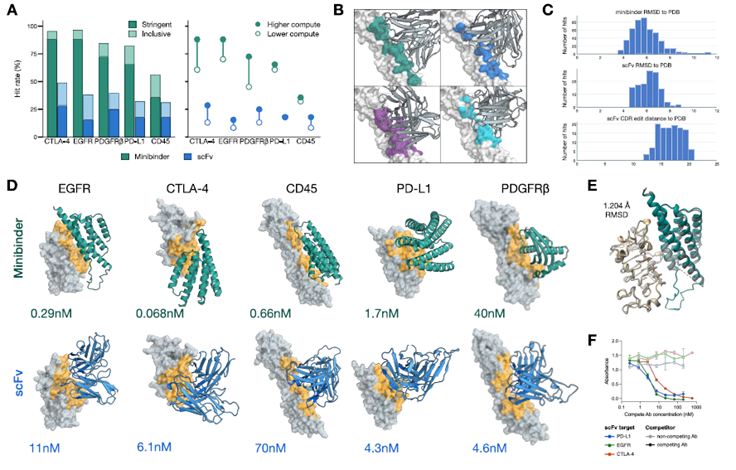
4. Validación de Laboratorio Húmedo: Éxito Masivo de los “Minibinders” 🧫🔬
Para validar el modelo, los científicos utilizaron un procedimiento de búsqueda simple para diseñar computacionalmente nuevas proteínas capaces de unirse a dianas terapéuticas reales de enfermedades humanas:
- Las Dianas del Cáncer: Se diseñaron aglutinantes (binders) contra receptores celulares críticos: EGFR, PDGFRβ, PD-L1, CTLA-4 y CD45.
- Resultados Experimentales: Al sintetizar físicamente los diseños en el laboratorio, se obtuvieron tasas de éxito masivas de entre el 36% y el 88% para las miniproteínas de diseño, alcanzando afinidades nanomolares muy potentes. Al escalar el poder de cómputo durante la fase de diseño, la tasa de éxito saltó del 54% al 70%, demostrando que a mayor capacidad de inferencia, mayor es la certeza del fármaco.

- Prueba Atómica por Cryo-EM: El paper documenta la resolución por Criomicroscopía Electrónica de uno de los complejos proteicos diseñados artificialmente unido al receptor EGFR humano, confirmando que la posición de los átomos en el mundo real era idéntica a la simulación digital de ESMFold2.

5. El ESM Atlas y el Descubrimiento de CRISPRs Eucariotas 🗺️🍄
El despliegue informático de este trabajo cristalizó en el ESM Atlas, que con sus 1,100 millones de estructuras 3D expande dramáticamente el conocimiento de la diversidad molecular del planeta, enfocándose en la “materia oscura” metagenómica.
- Buscando Agujas en un Pajar Metagenómico: Gracias a que la base de datos incluye secuencias que jamás han sido cultivadas en un laboratorio, los investigadores usaron herramientas de búsqueda estructural para rastrear familias de proteínas homólogas.

- El Hallazgo Sorpresa: Encontraron que proteínas del sistema de defensa bacteriano CRISPR comparten una arquitectura tridimensional casi idéntica con una proteína de edición genética descubierta recientemente en un hongo del suelo y compartida por otros organismos eucariotas. Esto demuestra que el Atlas sirve para rastrear cómo las herramientas biológicas saltaron entre reinos a lo largo de la evolución.
📚 REFERENCIA BIBLIOGRÁFICA
- Candido, S., Hayes, T., Derry, A., Rao, R., Lin, Z., Wu, B., Lee, J. S., Bruguera, E. S., Keval, J. A., Kopylov, M., Wu, W., Thomas, N., Mataraso, S., Hsu, A., Trotman-Grant, A., Verkuil, R., Pak, J. E., Fatras, K., Deaton, J., dos Santos Costa, A., Badkundri, R., Akın, H., Oktay, D., Montabana, E., Sitwala, H., Yu, Y., Wiggert, M., Carlin, D. A., Goering, A. W., Blazejewski, T., Sandora, M., Hla, M., Jia, T. Z., Kloker, L. H., Sofroniew, N. J., Uehara, M., Pannu, J., Bachas, S., Liu, D. S., Sercu, T., & Rives, A. (2026). Language Modeling Materializes a World Model of Protein Biology. Chan Zuckerberg Biohub / EvolutionaryScale Preprint, May 2026. https://biohub.ai/esm/protein/