En Pocas Palabras:
Nuestro ADN contiene la información para fabricar proteínas, pero el proceso desde el gen hasta la proteína funcional es increíblemente complejo e involucra muchas variaciones (edición de ARN, splicing alternativo, fusiones génicas, etc.). Esto significa que se producen muchas proteínas “no canónicas” que no están en las bases de datos de referencia. Detectarlas es un gran desafío, pero crucial para entender enfermedades como el cáncer. Un nuevo estudio presenta moPepGen, un algoritmo basado en grafos que genera de manera exhaustiva y eficiente listas de estos péptidos no canónicos a partir de datos genómicos y transcriptómicos. moPepGen supera a las herramientas existentes en cobertura y precisión, permitiendo identificar péptidos previamente invisibles derivados de variantes genéticas, regiones no codificantes e incluso ARN circular, abriendo nuevas vías para la proteogenómica y el descubrimiento de biomarcadores.
El Vasto y Oculto Mundo de las Proteínas No Canónicas
¡GRACIAS POR LEER NUESTRAS NOTICIAS! ¿Nos invitas un cáfe? ☕

El dogma central de la biología molecular nos dice que el ADN se transcribe en ARN y este se traduce en proteínas, las moléculas que realizan la mayoría de las funciones en nuestras células. Sin embargo, esta es una visión simplificada. Un solo gen puede dar lugar a múltiples productos proteicos a través de una miríada de procesos complejos como la variación genética (mutaciones), el splicing alternativo del ARN (donde diferentes partes de un gen se unen de distintas maneras), la edición del ARN (cambios químicos en la secuencia del ARN), las fusiones génicas (cuando partes de dos genes diferentes se unen) e incluso la circularización del ARN. El resultado es un universo de proteínas y péptidos “no canónicos” que no se encuentran en las bases de datos de referencia estándar, pero que pueden ser cruciales para entender la biología celular normal y, especialmente, enfermedades complejas como el cáncer. Identificar estos péptidos “oscuros” es un desafío computacional enorme, pero esencial para avanzar en la proteogenómica (el estudio de las proteínas a gran escala directamente desde la información genómica y transcriptómica). Un nuevo y poderoso algoritmo llamado moPepGen, presentado en Nature Biotechnology por Chenghao Zhu, Paul C. Boutros, Thomas Kislinger y colaboradores, promete iluminar este “proteoma oscuro” de manera integral y eficiente.

El Desafío: Modelando la Complejidad de la Expresión Génica
Las estrategias actuales para detectar péptidos no canónicos, como la secuenciación de novo de espectros de masas o las búsquedas “abiertas”, son computacionalmente costosas, tienen altas tasas de falsos negativos y dificultan la identificación precisa de la variante que originó el péptido. La mayoría de los estudios de proteogenómica, por lo tanto, se basan en la creación de bases de datos de péptidos personalizadas que incorporan alteraciones de ADN y ARN específicas de la muestra que se está analizando. Sin embargo, modelar exhaustivamente la diversidad combinatoria de todos los posibles productos proteicos es un reto monumental. Las herramientas existentes a menudo se centran en tipos individuales de variantes o simplifican demasiado el proceso, perdiendo muchos péptidos no canónicos.
moPepGen: Un Enfoque Basado en Grafos para una Cobertura Exhaustiva
Para superar estas limitaciones, los investigadores desarrollaron moPepGen (generador de péptidos multi-ómicos). Este innovador algoritmo utiliza una estructura de datos basada en grafos para modelar de manera eficiente y exhaustiva todas las posibles secuencias de proteínas que pueden surgir de un transcrito dado, considerando cualquier combinación de:
- Variantes genómicas (SNPs, pequeños indels, tanto germinales como somáticos).
- Sitios de edición de ARN.
- Eventos de splicing alternativo (como intrones retenidos).
- Fusiones de transcritos.
- ARN circular (circRNA).
- Marcos de lectura abiertos (ORFs) en regiones no codificantes.

moPepGen construye un “grafo de variantes de transcrito”, luego un “grafo de variantes de péptido” y finalmente un “grafo de clivaje de péptido”, permitiendo un recorrido sistemático a través de todas las combinaciones posibles en un tiempo lineal, lo cual es una mejora drástica en eficiencia computacional respecto a los métodos de fuerza bruta.
Validación y Poder de Descubrimiento
Los autores demostraron la robustez, precisión y poder de moPepGen en diversos conjuntos de datos:
- Precisión y Eficiencia: En pruebas de simulación (“fuzz testing”), moPepGen demostró una precisión perfecta y una complejidad de tiempo lineal, superando drásticamente a los enfoques de fuerza bruta.
- Mayor Cobertura que Herramientas Existentes: Al compararlo con otras herramientas populares para generar bases de datos personalizadas (como customProDB y pyQUILTS) en datos reales de cáncer de próstata, moPepGen identificó un número significativamente mayor de péptidos no canónicos únicos, especialmente aquellos derivados de combinaciones complejas de variantes.
- Identificación de Péptidos en Múltiples Contextos:
- Variación Germinal: En proteomas de ratón, moPepGen predijo e identificó péptidos no canónicos derivados de SNPs e indels específicos de la cepa.
- Mutaciones Somáticas en Cáncer: En un análisis de 375 líneas celulares de cáncer, moPepGen generó bases de datos de variantes en minutos por línea celular e identificó numerosos péptidos derivados de mutaciones somáticas y fusiones génicas, incluyendo aquellas en genes conductores del cáncer como KRAS y TP53.
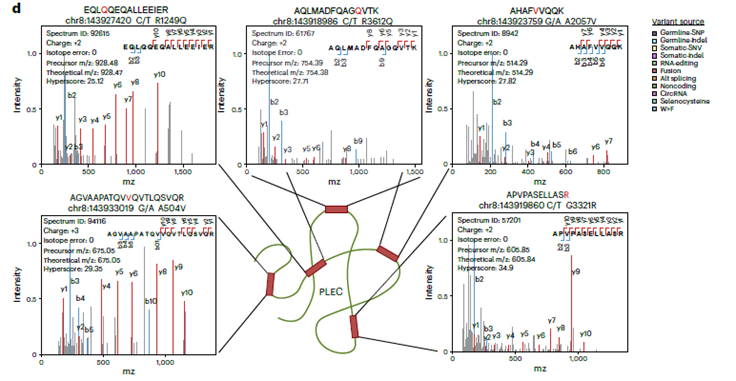
- Eventos de ARN Complejos: En muestras de cáncer de próstata primario con datos genómicos y transcriptómicos profundos, moPepGen identificó péptidos derivados no solo de SNPs y splicing alternativo, sino también de ARN circular y de ORFs en regiones previamente consideradas no codificantes, incluyendo algunas con modificaciones post-traduccionales como la sustitución de triptófano por fenilalanina.
Implicaciones para la Proteogenómica y la Medicina de Precisión
moPepGen tiene el potencial de transformar el campo de la proteogenómica:
- Descubrimiento Expandido: Permite una exploración mucho más profunda y completa del proteoma no canónico, revelando péptidos que antes eran invisibles.
- Mejora en la Identificación de Neoantígenos: Al identificar con mayor precisión péptidos derivados de mutaciones somáticas, puede mejorar la búsqueda de neoantígenos, que son dianas cruciales para la inmunoterapia contra el cáncer.
- Entendimiento de la Biología del Cáncer: La capacidad de detectar péptidos de fusiones génicas, ARN circular y ORFs no codificantes puede arrojar nueva luz sobre los mecanismos moleculares del cáncer y la desregulación de la expresión génica.
- Herramienta Universal: moPepGen es versátil, funciona con múltiples tecnologías de secuenciación, en diferentes especies y con todo tipo de datos genéticos y transcriptómicos.
- Flujos de Trabajo Simplificados: Se integra en flujos de trabajo proteómicos existentes y está diseñado para ser accesible, con una implementación en Python y un pipeline de Nextflow para automatizar el preprocesamiento de datos.

Conclusión: Iluminando la “Jungla” de las Proteínas No Convencionales
El desarrollo de moPepGen por Zhu y sus colegas representa un avance significativo en nuestra capacidad para explorar la vasta y compleja diversidad del proteoma. Al proporcionar una herramienta computacionalmente eficiente y exhaustiva para generar péptidos no canónicos a partir de la integración de datos multi-ómicos, moPepGen no solo supera muchas de las limitaciones de los métodos actuales, sino que también abre la puerta a nuevos descubrimientos en la biología fundamental, la investigación del cáncer y la medicina personalizada. Es como si nos hubieran dado un mapa mucho más detallado y una linterna más potente para explorar la intrincada “jungla” de las proteínas que realmente existen en nuestras células.
Referencia del Artículo:
Zhu, C., Liu, L. Y., Ha, A., Yamaguchi, T. N., Zhu, H., Hugh-White, R., … & Boutros, P. C. (2025). Identification of non-canonical peptides with moPepGen. Nature Biotechnology. https://doi.org/10.1038/s41587-025-02701-0
(Publicado online: 16 de junio de 2025).