Resumen 💡
La microscopía de luz convencional siempre ha estado limitada por la difracción, lo que impide observar la estructura fina de las proteínas a escala subnanométrica sin recurrir a complejos métodos de superresolución óptica o a la criomicroscopía electrónica. En este trabajo se presenta 1000ExM (Microscopía de Expansión de Mil Aumentos), una revolucionaria técnica basada en un gel de polímeros interpenetrados de cuatro redes secuenciales cargadas iónicamente. Este método logra una expansión lineal de aproximadamente 1000 veces (un billón de veces en volumen), separando físicamente los aminoácidos individuales de las proteínas. Al estirar las distancias intermoleculares por encima del límite de difracción, es posible mapear la estructura tridimensional de péptidos y proteínas con resolución de aminoácidos mediante microscopios de fluorescencia estándar. Las simulaciones bioinformáticas demuestran que estas coordenadas espaciales 3D permiten identificar de manera única más del 99% de las proteínas del proteoma humano, lo que abre la puerta a la identificación proteica in situ, directamente en células y tejidos.

1. El Desafío Científico y el Límite de Difracción 🎯
¡GRACIAS POR LEER NUESTRAS NOTICIAS! ¿Nos invitas un cáfe? ☕

- La barrera física: Los microscopios ópticos convencionales tienen un límite de difracción lateral de 200 a 300 nm. Las distancias entre carbonos alfa adyacentes en el esqueleto de una proteína son de apenas ~0.38 nm.
- La hipótesis: Si se anclan las cadenas laterales de los aminoácidos a una matriz polimérica hidrofílica y expandible, y luego se corta el esqueleto peptídico, es posible separar físicamente los aminoácidos a distancias microscópicas. Con una ampliación de 1000x, los ~0.38 nm originales se transforman en ~380 nm, un rango perfectamente resoluble con microscopía confocal estándar.

2. Innovación Química: Geles Iónicos Interpenetrados sin Reembebido Neutro 🧪
- Rompiendo el paradigma: Los métodos tradicionales de microscopía de expansión iterativa (como iExM) utilizan geles neutros intermedios para estabilizar la matriz antes de la siguiente ronda, lo que limita la expansión a 16-25x debido a la restricción mecánica.
- La arquitectura 1000ExM: Se diseñó una secuencia de cuatro redes poliméricas iónicas basadas en una formulación optimizada de acrilato de sodio y N,N-dimetilacrilamida (SA/DMAA).
- Progresión geométrica de expansión:
- Ronda 1: Inclusión del espécimen y digestión proteica à Expansión lineal de ~18x.
- Ronda 2: Infiltración con solución de monómeros cargados (contracción temporal a ~10x) y polimerización àExpansión a ~100x.
- Ronda 3: Repetición del proceso iónico-en-iónico à Expansión a ~500x.
- Ronda 4: Última polimerización y expansión en agua purificada → Máxima expansión de ~1500x, estabilizada en PBS a ~1000x para la adquisición.
c
3. Validación Métrica con Reglas Moleculares 📐
- Escala nanométrica (Nanoanticuerpos): Se utilizaron nanoanticuerpos con dos fluoróforos separados entre sí por ~4 nm. A lo largo de todas las rondas de expansión, las distancias calculadas se mantuvieron proporcionales e isótropas, lo que igualó las proyecciones moleculares del cristal original.
- Escala subnanométrica (Péptido mCLING): Al expandir el péptido mCLING a 1000x, los picos de señal fluorescente de los aminoácidos individuales se separaron con una precisión extraordinaria. El análisis 3D de 6,284 péptidos individuales mostró una desviación de la raíz cuadrada media (RMSD) de apenas 0.573 ± 0.151 nm frente a modelos de dinámica molecular.

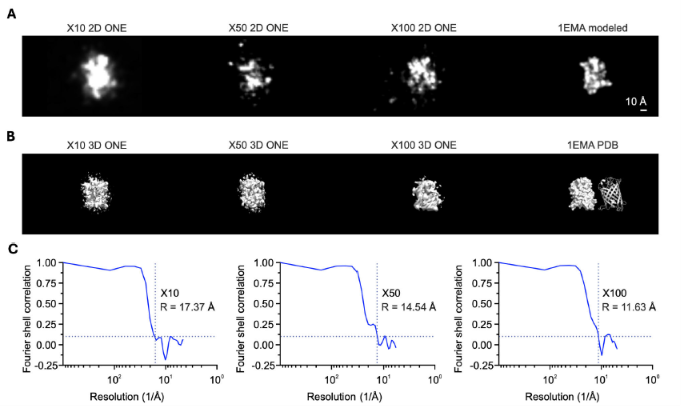
4. Reconstrucción 3D de Proteínas Complejas (GFP) 🟢
- Al evaluar la Proteína Verde Fluorescente (GFP) mediante la combinación de expansión (100x) y microscopía ONE, se logró una resolución estructural de 11.63 Å verificada por Correlación de Concha de Fourier (FSC). Esto se acerca al límite fundamental de muestreo químico impuesto por la distancia de las cadenas laterales de las lisinas ancladas.

5. Identificación Computacional del Proteoma Humano 💻
- Se simuló la viabilidad de identificar 23,391 proteínas humanas basándose únicamente en las coordenadas tridimensionales de sus residuos superficiales expuestos bajo la distorsión experimental medida de 1000ExM.

- Poder de discriminación cromática:
| Clases de Aminoácidos Marcados | Colores / Canales | Identificabilidad del Proteoma (Distorsión 5 Å) | Identificabilidad del Proteoma (Distorsión 10 Å) |
| Solo Lisina | 1 color | 89.3 – 93.3% | 78.0 – 83.8% |
| Lisina + Cisteína | 2 colores | 93.9 – 96.5% | 84.2 – 89.2% |
| Lisina + Cisteína + Aspartato/Glutamato | 3 colores | > 99.3% | 96.4 – 97.8% |
Como puede verse en este trabajo, esperando que la revisión por pares valide el trabajo, esta nueva técnica aumenta el poder de resolución de la microscopía de luz y ahora se podrá evaluar la configuración espacial de proteínas.
Referencia 📚
- [Preprint de Origen] Hu, H., Krah, D., Ntolkeras, A., Chanda, S., Heimbrodt, A., Mondal, M., Altendorf, J., Jing, B., Berger, B., Shaib, A. H., Rizzoli, S. O., & Boyden, E. S. (2026). Thousandfold Expansion Microscopy. bioRxiv preprint doi: https://doi.org/10.64898/2026.05.31.729018.